Conference
Secretariat:
Conferences and Events Ltd
PO Box 24078, Manners St,
Wellington 6142
Email:
[email protected]
Tel: +64 4 384 1511
Fax: +64 4 384 4667
|

Speaker Abstracts
Details
of the speaker Abstracts will be posted as they become available.
Daniel
Nocera (Massachusetts Institute of Technology)
Personalized
Energy for 1 (� 6 Billion)
Abstract. The capture and storage of solar energy at the individual
level – personalized solar energy – drives inextricably towards the
heart of this energy challenge by addressing the triumvirate of secure,
carbon neutral and plentiful energy. Because energy use scales with
wealth, point-of-use solar energy will put individuals, in the smallest
village in the non-legacy world and in the largest city of the legacy
world, on a more level playing field. Moreover, personalized energy
(PE) is secure because it is highly distributed and the individual
controls the energy on which she/he lives. Finally, the doubling of
global energy need by mid-century and tripling by 2100 is driven by 3
billion low-energy users in the non-legacy world and by 3 billion
people yet to inhabit the planet over the next half century. The
possibility of generating terawatts of carbon-free energy, and thus
providing society with its most direct path to realizing a low GHG
future, may be realized by making solar PE available to the 6 billion
new energy users by high throughput manufacturing. Notwithstanding,
current options to harness and store solar energy at the individual
level are too expensive to be implemented, especially in a non-legacy
world. The imperative to science is to develop new materials, reactions
and processes that enable personalized solar energy to be sufficiently
inexpensive to penetrate global energy markets and especially the
non-legacy world.
Personalized energy at low cost presents new basic research targets.
Because personalized energy will be possible only if solar energy is a
24/7 available supply, the key enabler for personalized energy is
inexpensive storage. Studies in the Nocera group have led to the
creation of a new catalyst that captures many of the functional
elements of photosynthesis and in doing so provides a highly
manufacturable and inexpensive method to effect a carbon-neutral and
sustainable method for solar storage – solar fuels from
water-splitting. By developing an inexpensive 24/7 solar energy system
for the individual, a carbon-neutral energy supply for 1 � 6 billion
becomes available.
Back
Prof Akihiko
Kudo Department of Applied
Chemistry, Tokyo University of Science,
1-3 Kagurazaka, Shinjuku-ku, Tokyo 162-8601 Japan.
E-mail address: [email protected]
Photocatalysts
for solar hydrogen production
The
importance of hydrogen energy has recently been re-recognized because
of the interest in clean energy. Hydrogen must be produced from water
using a renewable energy source, if one considers the energy and
environmental issues. Therefore, photocatalytic water splitting is a
challenging reaction because it is an ultimate solution to these
serious problems. In the present paper, we introduce various metal
oxide and sulfide photocatalysts aiming at water splitting [1]. Many
visible-light-driven photocatalysts have been developed through band
engineering by doping of metal cations, new valence formation, and by
making solid solution. Among them, Ru/SrTiO3 doped with Rh showed high
activity for H2 evolution from aqueous solutions containing a reducing
reagent under visible light irradiation. BiVO4 showed high activity for
O2 evolution in the presence of sacrificial reagent (Ag+). Overall
water splitting under visible light irradiation has been achieved by
construction of a Z-scheme photocatalysis system employing these
visible-light-driven photocatalysts (Ru/SrTiO3:Rh and BiVO4) and an
Fe3+/ Fe2+ redox couple as an electron mediator. Moreover, Z-scheme
photocatalysis system consisting of Ru/SrTiO3:Rh and BiVO4 without the
electron mediator showed activity for water splitting when pH was
adjusted at 3.5. These Z-scheme systems with and without an electron
mediator were active for solar water splitting. Although the photon
energy conversion using powdered photocatalysts is not at the stage of
practical use, the research in photocatalytic water splitting is being
advanced.
References
[1] A. Kudo and Y. Miseki, Chem. Soc. Rev., 2009, 38, 253-278.
Back
Professor James K. McCusker
Department of Chemistry, Michigan State University
Ultrafast Excited-state Processes in Transition Metal-based Chromophores:
From Fundamental Photophysics to Applications in Energy Science
Recent
attention to the dangers of climate change has spurred renewed efforts
toward developing carbon-neutral sources of energy. With global energy
needs expected to at least double by 2050, the urgency of finding
alternatives to fossil fuels is very real. Although there is no “silver
bullet” for this impending crisis – all possible avenues must be
explored – solar energy stands out as one option that has perhaps the
greatest potential in terms of worldwide application. At present, one
of the fundamental problems associated with large-scale implementation
of solar-based energy technologies is cost. In 1991, O’Regan and
Gr�tzel published a report that heralded a new direction in the
development of low-cost photovoltaics. By exploiting 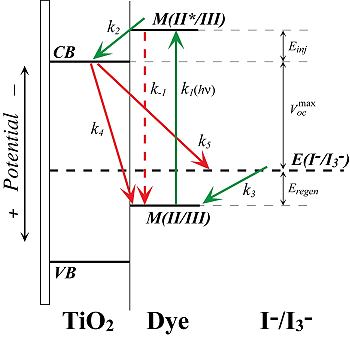 the
concept of dye-sensitization of semiconductors, these workers
fabricated a solar cell based on a mesoporous film of nanocrystalline
TiO2. In conjunction with a molecular chromophore, these devices have
the potential to form the basis of photoconversion strategies that
could make solar energy economically competitive. In order to realize
this technological goal, however, there are a number of scientific
issues that must be addressed and ultimately overcome. the
concept of dye-sensitization of semiconductors, these workers
fabricated a solar cell based on a mesoporous film of nanocrystalline
TiO2. In conjunction with a molecular chromophore, these devices have
the potential to form the basis of photoconversion strategies that
could make solar energy economically competitive. In order to realize
this technological goal, however, there are a number of scientific
issues that must be addressed and ultimately overcome.
This seminar
will highlight our efforts to develop semiconductor-based
dye-sensitized photovoltaics based on chromophores involving first-row
transition metal complexes. The motivation for this line of research
stems largely from the lower cost and greater scalability associated
with these materials as opposed to the second- and third-row complexes
currently being employed. In the course of our research, we have
discovered that differences in electronic structure endemic to
first-row versus isoelectronic second- or third-row complexes give rise
to a fundamental change in the excited-state dynamics of such compounds
that directly impacts the ability to incorporate this class of
molecules into this technology. The key experimental findings
establishing this paradigm will be described, along with strategies
that we are currently pursuing to circumvent these problems in order to
realize cheaper, more efficient photovoltaic devices.
Back
Cather
Simpson (University of Auckland)
A New Twist in the Tale: Ultrafast Dynamics of Diphosphenes and Phosphaalkenes
Diphosphenes
are a relatively new class of molecules that contain a central –P=P–
bond stabilized by bulky protecting groups. As the chemical
cousins of stilbenes (-C=C-) and azobenzenes (-N=N-), diphosphenes have
potential for use as photoactive elements in photonic devices.
However, while a great deal is known about the photobehaviour of
stilbenes and azobenzenes, virtually no detailed information about the
photophysics and photochemistry of molecules with heavier main group
double bonds, like diphosphenes, has been reported. Here we
present the first spectroscopic observations of the ultrafast dynamics
of these molecules. Femtosecond transient absorption spectroscopy
(fsTRA) of the excited states of diphosphenes in solution (~100 fs
resolution) has been used in conjunction with high-level quantum
mechanical calculations (CASPT2//CASSCF) to explore the response of
diphosphenes to photoexcitation in resonance with the low-lying n-*,
the -*, and the higher-lying * states. The lifetimes of the
transient intermediate states are very short, with the dynamics
complete in about a nanosecond. The presence of conical
intersections in the computational findings – and the absence of
significant fluorescence emission – supports these rapid electronic
state interconversions. Intriguingly, preliminary results
indicate that the triplet manifold may play an important role in
diphosphene photochemistry. Such behaviour would represent a
significant departure from the all-singlet paradigm of stilbene and
azobenzene photochemistry.
Back
Hiroshi Miyasaka, Tetsuro Katayama, Yukihide Ishibashi
Graduate School of Engineering Science, Center for Quantum Science and
Technology under Extreme Conditions, Osaka University, and CREST,
Toyonaka, Osaka 560-8531, Japan
Femtosecond Delocalization Dynamics of Cationic States in Photoconductive Poly(N-vinylcarbazole) Amorphous Solid.Poly(N-vinylcarbazole) (PVCz) is one of the most well-known organic photoconductive materials. PVCz and its analogues are still regarded as representative references and widely utilized as building units in advanced functional systems. A scheme based on the Onsager model has been conventionally employedfor interpretation of primary processes in the photoconduction, where the key process is rapid increase in the interionic distance during thermalization (ca. 10 ps) after photoinduced charge separation (CS).On the other hand, direct detection by picosecond transient dichroism measurements revealed that the hole escapes from the initial ion pair with a time constant of 1 ns, in competition with geminate charge recombination in the initial charge-separated state, followed by the sequential hole hopping process. Clearelucidation of these two conflicting models described above is quite important for the comprehensive understanding of the carrier photo-generation phenomenon and for rationally designing advanced organic photoconductive materials and other systems related to electron transport.Along this line, we have investigated the photoprimary processes in PVCz solid films doped with an electron acceptor by means of femtosecond dichroism measurement in the visible region and of transient absorption spectroscopy in the near-infrared (NIR) region. From these measurements, it was observed that the cationic state of the carbazolyl group undergoes very rapid delocalization in sub-ps to several ps time region, leading to the reduction of the Coulombic attraction between the cation and the counter anion. The role of the delocalization in the subsequent hole migration reaction will be discussed in detail.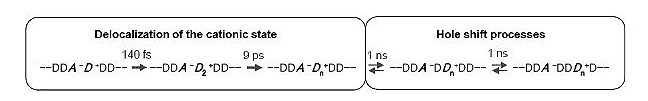 Scheme 1. Reaction scheme for the electron transfer processes in the photoconductive PVCz solid film doped with an electron acceptor after photoinduced charge separation between A and D. Here, D is a carbazolyl unit (an electron donor) and A is an electron acceptor. Charge recombination processes are not written in the scheme. Typical value of n, which is the number of the D unit in the delocalized cation, is ≥ 3. Scheme 1. Reaction scheme for the electron transfer processes in the photoconductive PVCz solid film doped with an electron acceptor after photoinduced charge separation between A and D. Here, D is a carbazolyl unit (an electron donor) and A is an electron acceptor. Charge recombination processes are not written in the scheme. Typical value of n, which is the number of the D unit in the delocalized cation, is ≥ 3.
Guoqiang Yang Key Laboratory of Photochemistry, Institute of Chemistry,
Chinese Academy of Sciences, Beijing 100190, P. R. China. Email: [email protected]
Photoluminescent materials: structure, property and high pressure effect
Excited
state intramolecular proton transfer (ESIPT) compounds and
intramolecular charge transfer (ICT) compounds have attracted much
attention for their luminescent properties. The luminescent
characteristics of the compounds are sensitive to the environment,
include the high pressure. For the ESIPT compounds, a fast four-level
photophysical cycle occurs immediately after photo-excitation. The
emission from the proton transfer state gives abnormally large Stokes
shift and no self-absorption is detected. For the ICT compounds, the
emission shows red shift with the increasing of the solvent polarity.
Meanwhile, significant changes of the luminescent properties are
observed from solution to aggregation. For the good photo-stability and
unique luminescent properties, ESIPT compounds and ICT compounds are
expected to be potential intrinsic luminescent materials and could be
used as special probes for small molecules and metallic ions.
References:
- Rui
Hu, Jiao Feng, Dehui Hu, Shuangqing Wang, Shayu Li,Yi Li and Guoqiang
Yang, Angew. Chem. Int. Ed., 49 (29), 4915-4918 (2010).
- Wenhao Sun, Shayu Li, Rui Hu, Yan Qian, Shuangqing Wang, Guoqiang Yang,J. Phys. Chem. A, 113, 5888–5895 (2009).
- Xiuping Li, Yan Qian, Shuangqing Wang, Shayu Li, Guoqiang Yang, J. Phys. Chem. C, 113 (9), 3862-3868 (2009).
- Qian
Wang, Shayu Li, Liming He, Yan Qian, Xiuping Li, Wenhao Sun, Min Liu,
Juan Li, Yi Li, Guoqiang Yang , ChemPhysChem, 9, 1146-1152(2008).
- Yan
Qian, Shayu Li, Guoqi Zhang, Qian Wang, Shuangqing Wang, Huijun Xu,
Chengzhang Li, Yi Li, Guoqiang Yang, J. Phys. Chem. B, 111,
5861-5868(2007).
- Shayu Li, Qian Wang, Yan Qian, Shuangqing Wang, Yi Li, Guoqiang Yang, J. Phys. Chem. A, 111, 11793-11800 (2007).
- Shayu Li, Liming He, Fei Xiong, Yi Li, Guoqiang Yang, J. Phys. Chem., B, 108(30), 10887(2004)
Back
Keith Millington (Australian Commonwealth Scientific and Research Organization)
Studying the photodegradation of materials using chemiluminescence
Keith R. Millington 1, Michael J. Jones 1 and Siti Farhana Zakaria 2
1 CSIRO Materials Science and Engineering, Belmont, VIC 3216, Australia.
2 School of Fashion and Textiles, RMIT University, Brunswick, VIC 3056, Australia.
Polymers
and organic materials that are exposed to sunlight undergo
photooxidation, which leads to deterioration of their physical
properties. To allow adequate performance under outdoor conditions,
synthetic polymers require additives such as antioxidants and UV
absorbers. A major problem with optimising polymer formulations to
maximise their working lifetime is that accelerated weathering tests
are empirical. The conditions differ significantly from real weathering
situations, and samples require lengthy irradiation times. No
degradation may be apparent in the early stages of exposure, although
this is when products such as hydroperoxides are formed which later
cause acceleration of oxidation.
Chemiluminescence (CL) has
been widely used to study the thermal oxidation of organic materials.
CL originates from side reactions of peroxy radical and hydroperoxide
intermediates formed during early stages of the autoxidation chain
reaction. However the application of CL to study the photodegradation
of materials has been very limited. We describe a simple modification
to a commercial CL instrument (Lumipol 3) and an effective experimental
protocol to study the photo-induced chemiluminescence (PICL) from
irradiated materials. The CL instrument was reversibly modified to
allow in situ irradiation with selected wavelengths from a
medium-pressure mercury arc via a liquid light pipe, as shown in Figure
1.
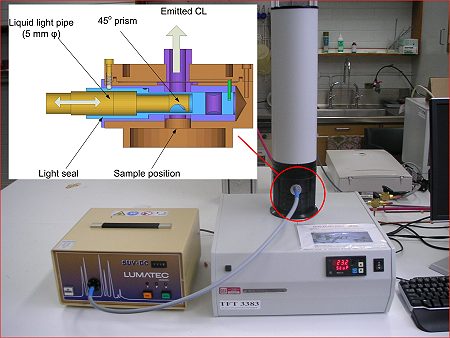
Figure
1. Lumipol CL instrument linked to Lumatec light source for PICL
studies. Inset shows the adaptor which allows samples to be irradiated
with wavelengths above 320 nm.
We have applied the PICL
technique to polymer films and coatings, fibrous webs, such as textile
fabrics or paper, and powdered samples, and the effects of additives,
dyes and pigments on the rate of oxidation can also be assessed.
Back
Kenneth
Kam-Wing Lo Department of
Biology and Chemistry, City University of Hong
Kong, Tat Chee Avenue, Kowloon, Hong Kong, P. R. China.
E-mail:
[email protected]
Luminescence
and Biological Properties of New Cyclometalated Iridium(III)
Polypyridine Complexes
Many
cyclometalated iridium(III) polypyridine complexes exhibit intense and
long-lived emission that is very sensitive to the molecular structures
and local environments of the complexes. These interesting
properties allow the complexes to serve as useful probes for various
biological molecules including oligonucleotides, peptides, and
proteins. We have attached amine- and sulfhydryl-specific
reactive functional groups such as isothiocyanate, aldehyde, and
iodoacetamide to cyclometalated iridium(III) polypyridine complexes of
the type [Ir(N^C)2(N^N)]+ to yield new luminescent labels for
biomolecules. Additionally, we have designed related
iridium(III)
polypyridine complexes appended with various biological substrates
including indole, -estradiol, biotin, and lipids, and utilized the
complexes as luminescent probes for indole-binding proteins, estrogen
receptors, avidin, and lipid-binding proteins, respectively.
Some
of these complexes show interesting dual-emissive properties that
enable the biological binding event to be reflected by a change of
emission profiles of the probes. Furthermore, we have
recently
developed DNA-metallointercalators, dendrimers, and PEGylation reagents
derived from luminescent iridium(III) polypyridine complexes.
We
have focused on the molecular design, photophysical properties,
biomolecule-binding behavior, cytotoxicity, and cellular-uptake
characteristics of these luminescent probes.
- Lo, K.
K.-W.; Zhang, K. Y.; Chung, C.-K.; Kwok, K. Y. Chem. Eur. J. 2007, 13,
7110 – 7130.
-
Lo, K. K.-W.; Zhang, K. Y.; Leung,
S.-K.; Tang, M.-C. Angew. Chem. Int. Ed. 2008, 47, 2213 – 2216.
-
Zhang, K. Y.; Li, S. P.-Y.; Zhu, N.; Or, I. W.-S.; Cheung, M. S.-H.;
Lam, Y.-W.; Lo, K. K.-W. Inorg. Chem. 2010, 49, 2530 – 2540.
- Zhang, K. Y.; Liu, H.-W.; Fong, T.
T.-H.; Chen, X.-G.; Lo, K. K.-W. Inorg. Chem. 2010, in press.
-
Li, S. P.-Y.; Liu, H.-W.; Zhang, K.
Y.; Lo, K. K.-W. Chem. Eur. J. 2010, in press.
Back
Chi-Kung Kenny Ni
Institute of Atomic and Molecular Sciences, Academia Sinica, Taipei, Taiwan
Molecular mechansim on the photostability of amino acid chromophores
Aromatic
amino acids like tryptophan and tyrosine have very large UV absorption
cross-sections and low fluorescence quantum yields. It indicates the
existence of fast nonradiative processes, which efficiently quench the
fluorescence. The nonradiative process was assumed to be the ultrafast
internal conversion. This so-called photostability prevents the
undesired photochemical reactions of these molecules upon the
irradiation of UV photons. Recent theoretical calculations suggest that
the low fluorescence quantum yields for indole and phenol, the
chromophore for the amino acid tryptophan and tyrosine, are due to the
dissociative characteristic of the excited electronic state potential
energy surfaces, rather than the fast internal conversion to the ground
electronic state. Because dissociation from an excited state having a
repulsive potential energy surface is swift, quenching is incomplete
even in the condensed phase. As a result, dissociation from the
repulsive state and the reactions following the generation of radicals
from dissociation become a potential problem in the photostability of
amino acids. We investigated the photodissociation properties of
various amino acid chromophores in a molecular beams using multimass
ion imaging techniques. We confirmed the dissociation from the
repulsive state for small amino acid chromophores. However, we
demonstrated that internal conversion becomes the dominant channel for
large chromophores. In addition, intramolecular hydrogen bonding was
found to play an important role to quench the dissociation from the
repulsive state. These observations provide an alternative explanation
on the photostability of amino acid chromophores.
Kyung Byung Yoon from Sogang University, Korea
Photovoltaic Effects of Zeolite-Encapsulated CdS and PbS Quantum Dots
Hyun Sung Kim, Nak Cheon Jeong, and Kyung Byung Yoon`
Korea Center for Artificial Synthesis, Department of Chemistry, Sogang University, Seoul 121-742, Korea
The
CdS and PbS quantum dot-incorporating zeolite Y films [(CdS)nYf and
(PbS)nYf, where, n denotes the loaded amount in %, 4.0. 5.3, 6.8 in the
case of CdS and 4.9, 6.9, and 11.0, in the case of PbS] supported on
ITO glass, respectively, showed photovoltaic effects in an electrolyte
solution composed of Na2S (1 M) and NaOH (0.1 M) with a Pt coated ITO
glass plate as the counter electrode and between themselves. Their
short circuit currents (isc), open circuit voltages (Voc), fill factors
(ff), and overall efficiencies (η) under the AM 1.5, 100 mW cm-2
condition (1 sun) were 0.3 mA cm�2, 423 mV, 28, 0.1% in the case of
(CdS)nYf/Pt pair and -0.15 mA cm�2, -58 mV, 29, 0.01% in the case of
(PbS)nYf/Pt pair, and the values significantly increased to 0.98 mA
cm�2, -478 mV, 30, 0.3% from the (CdS)nYf/(PbS)nYf couple. The maximum
IPCE and APCE values observed from (CdS)nYf/Pt were 14.3 and 30%,
respectively, at 390 nm while the IPCE value of (CdS)nYf/(PbS)nYf at
390 nm was 40%. The plot of the maximum APCE value with respect to the
loaded amount for (CdS)nYf/Pt showed that the APCE value increases in a
parabolic manner with respect to the loaded amount and the
extrapolation of the relationship predicted that the APCE value can
increase to the value higher than one at the loading level of 10%,
indicating that generation of multiple electrons from a single photon
could be realized by carefully optimizing the condition.
Back
George
Thomas (Indian Institute of Science Education and Research)
Optical Properties of Hybrid Nanomaterials
We
have recently initiated a detailed research program on the design of
nanoparticle conjugates of organic/inorganic molecules which enable the
coupling of the intrinsic functionalities of molecular systems
(binding, self-assembly, switching etc.) with the size and shape
dependent optoelectronic properties of nanomaterials.1 The presentation
will provide examples of modulating the optical properties of
nanomaterials by integrating them into higher order assemblies using
electrostatic, supramolecular and covalent approaches.2-11 The
presentation will also highlight our recent efforts to understand (i)
the interfacial properties of these hybrid nanomaterials, (ii) plasmon
coupling in hybrid metal nanostructures and (iii) the use of such
systems for Surface Enhanced Raman Spectroscopy (SERS).
Recent
studies from our group have shown that the organization of molecules
and nanomaterials on surfaces can be fine tuned by introducing proper
functional moieties and these aspects will be discussed.12-14 We have
recently developed a novel strategy for inducing chirality to metal
nanoparticle assembly by growing them on chiral surfaces having reduced
elements of symmetry. The surface plasmon coupled circular dichroism
observed in these systems originate from the asymmetric organization of
metal nanoparticles on surface resulting in bisignated CD signals.15
Mirror image relationship in the CD spectra indicates that the chiral
molecules on the D- and L- peptide nanotubes drive the organization of
nanoparticles in two different ways and these aspects will be discussed.
1.K. G Thomas, P. V. Kamat, Acc. Chem. Res. 2003, 36, 888.
2.K.
G. Thomas, chapter entitled “Surface plasmon resonances in
nanostructured materials,” in Nanomaterials chemistry: Novel aspects
and new directions, C.N.R. Rao, A. Mueller. A. K. Cheetham (Eds.)
Wiley-VCH (2007) pp 185-216.
3.S. T. S. Joseph, B. I. Ipe, P. Pramod, K. G. Thomas, J. Phys. Chem. B 2006, 110, 150.
4.P. K. Sudeep, S. T. S. Joseph, K. G. Thomas, J. Am. Chem. Soc. 2005, 127, 6517.
5.P. Pramod, S. T. S. Joseph, K. G. Thomas, J. Am. Chem. Soc. 2007, 129, 6712.
6.R. Vinayakan, T. Shanmugapriya, P. V. Nair, P. Ramamurthy, K. G. Thomas, J. Phys. Chem. C 2007, 111, 10146.
7.P. V. Nair, K. G. Thomas, J. Phys. Chem. Lett. 2010, 111, 2094.
8.B. I. Ipe, K. Yoosaf, K. G. Thomas, J. Am. Chem. Soc. 2006, 128, 1907.
9.K. Yoosaf, B. I. Ipe, C. H. Suresh, K. G. Thomas, J. Phys. Chem. C 2007, 111, 12839.
10.P. Pramod, K. G. Thomas, Adv. Mater. 2008, 20, 4300.
11.C. C. Soumya, P. Pramod, K. G. Thomas, Adv. Mater 2010 (submitted).
12.K. Yoosaf, P. V. James, A. R. Ramesh, C. H. Suresh, K. G. Thomas, J. Phys. Chem. C. 2007; 111, 14933.
13.K. Yoosaf, A. R. Ramesh, J. George, C. H. Suresh, K. G. Thomas, J. Phys. Chem. C. 2009, 113, 11836.
14.A. R. Ramesh, K. G. Thomas, Chem. Commun., 2010, 46, 3457.
15.J. George, K. G. Thomas, J. Am. Chem. Soc., 2010, 132, 2502.
Back
|
Sponsors


Bronze Sponsors




|
|
